Die Sichelzellkrankheit (SCD, sickle cell disease) ist eine Erbkrankheit, die durch eine Veränderung eines Hämoglobins durch eine einzige Basensubstitution im ersten Exon des β-Globin-Gens (HBB, Hämoglobin-Untereinheit beta) verursacht wird. Eine Punktmutation bewirkt einen Aminosäureaustausch der hydrophilen Aminosäure Glutaminsäure durch ein hydrophobe Aminosäure Valin im 7. Codon der β-Globinkette. Diese Substitution beeinträchtigt die Struktur und dadurch auch die Funktion des Hämoglobins erheblich und führt zu einer pathologischen Form des Hämoglobins, die als Sichelhämoglobin (α2βs2, HbS) bezeichnet wird.
Man fand Sichelzellanämie-Patienten, bei denen aufgrund einer davon unabhängigen Mutation im Genom fetales Hämoglobin (HbF) nach der Geburt nicht inaktiviert, sondern dauerhaft auch im Erwachsenenalter synthetisiert wird. Untersuchungen zeigten, dass diese Patienten aufgrund des erhöhten HbF-Anteils trotz Sichelzellanämie-Mutation fast keine Sichelzellanämie-Symptomatik aufweisen.
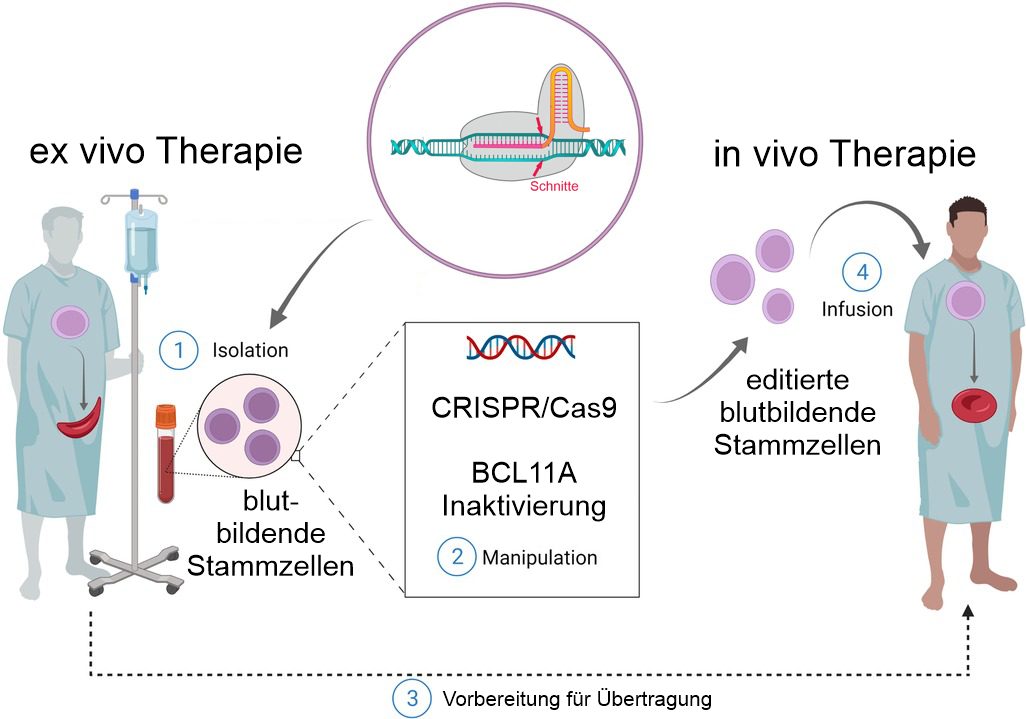
Neben Ansätzen zur Reparatur des HBB-Gens wurden deshalb bereits früh auch nach Wegen gesucht, durch medikamentöse oder gentechnische Eingriffe fetales Hämoglobin (HbF) dauerhaft zu exprimieren. CRISPR/Cas9 bietet durch Genom-Editing in blutbildenden Stammzellen für beide Wege Möglichkeiten.
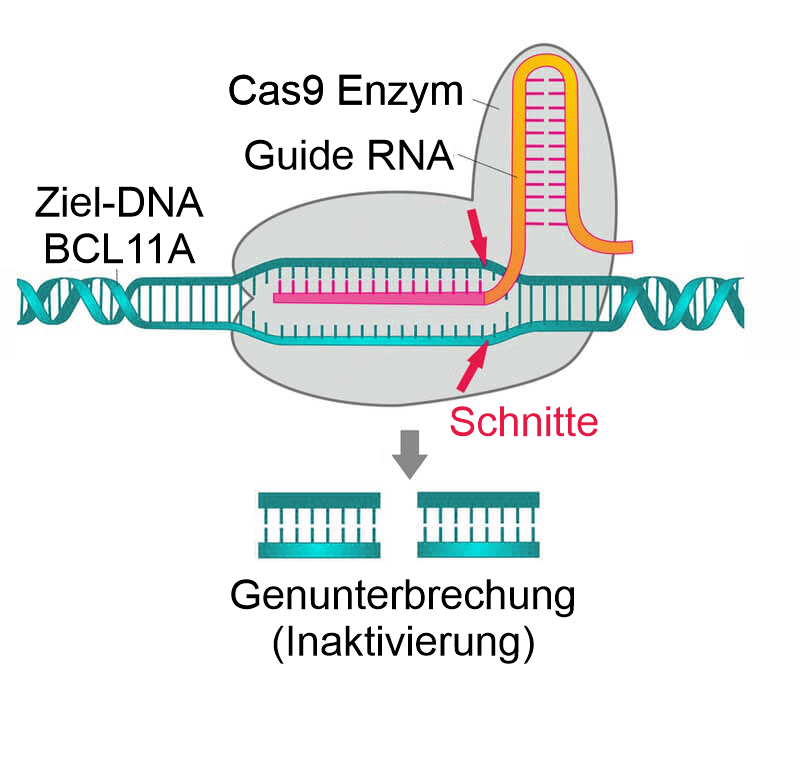
CRISPR/Cas9 schneidet und inaktiviert im aktuell zugelassen Verfahren das Gen BCL11A. Das von BCL11A codierte Protein ist ein Transkriptionsfaktor, der für die Unterdrückung der der HbF-Expression verantwortlich ist. Durch seine Inaktivierung kann HbF weiter transkribiert und fetales Hämoglobin gebildet werden.